set up notebook¶
In [1]:
#!rm genomes/tb927_6/*
In [2]:
#reload when modified
%load_ext autoreload
%autoreload 2
#activate r magic
%load_ext rpy2.ipython
%matplotlib inline
In [13]:
import os
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import utilities as UT
import missingno as msno
import random
import matplotlib.pyplot as plt
import matplotlib.patches as patches
import gc
random.seed(1976)
np.random.seed(1976)
Data Anaylsis¶
Experiment SetUp¶
In [14]:
from IPython.display import Image
In [ ]:
In [15]:
#create a dictionary of gene to desc
#from the gff file
def make_desc(_GFF):
gff =pd.read_csv( _GFF, sep='\t', header=None, comment='#')
gff = gff[gff.iloc[:,2]=='gene']
#print( gff[gff[gff.columns[-1]].str.contains('Tb427_020006200')] )
desc = {}
for n in gff.iloc[:,-1]:
n=n.replace('%2C',' ')
item_list = n.split(';')
#print (item_list)
temp_dict = {}
for m in item_list:
#print(m)
temp_dict[m.split('=')[0].strip()]=m.split('=')[1].strip()
#print(temp_dict['ID'])
#print(temp_dict['description'])
desc[temp_dict['ID']]=temp_dict.get('description','none')
return desc
desc_dict = make_desc('genomes/tb927_3/tb927_3.gff')
desc_dict['Tb10.v4.0073']
Out[15]:
In [16]:
exp = 'P{life_stage}{replica}'
list_df = [exp.format(
life_stage=life_stage,
replica=replica)
for life_stage in ['1','2','3']
for replica in ['C','T']
]
list_df = [n+'/res/'+n+'/counts.txt' for n in list_df]
list_df =[pd.read_csv(n,index_col=[0],comment='#',sep='\t') for n in list_df]
df = list_df[0].copy()
for temp_df in list_df[1:]:
df = df.join(temp_df.iloc[:,-1])
df.head()
#temp_df = pd.read_csv('BSF/tb927_3_ks_counts_final.txt',index_col=[0],comment='#',sep='\t')
Out[16]:
In [17]:
#df = pd.read_csv('InData/tb927_3_ks_counts_final.txt',index_col=[0],comment='#',sep='\t')
#df.head()
#data_col = df.columns[6:25]
In [18]:
data_col = df.columns[5:]
data_col
Out[18]:
In [19]:
indata = df[data_col]
indata.columns = [n.split('/')[3] for n in indata.columns]
indata = indata[['P1C','P2C','P3C','P1T','P2T','P3T']]
indata.head()
Out[19]:
In [20]:
print(indata.shape)
indata=indata.dropna()
print(indata.shape)
#indata.loc['KS17gene_1749a']
#indata['desc']=[desc_dict.get(n,'none') for n in indata.index.values]
#indata.to_csv('indata.csv')
#indata.head()
#indata.loc['mainVSG-427-2']
Missing Data Viz¶
In [ ]:
In [22]:
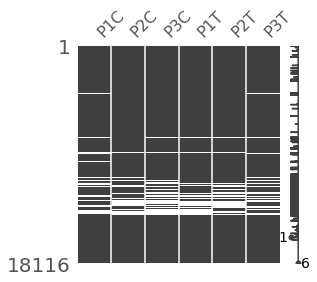
msno.matrix(indata.replace(0,np.nan).dropna(how='all'),figsize=(4, 4))
Out[22]:
In [23]:
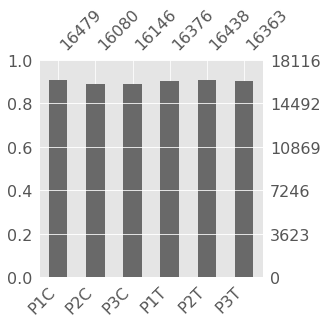
msno.bar(indata.replace(0,np.nan).dropna(how='all'),figsize=(4, 4))
Out[23]:
QC - Corr analysis¶
In [24]:
!mkdir -p Figures
In [27]:
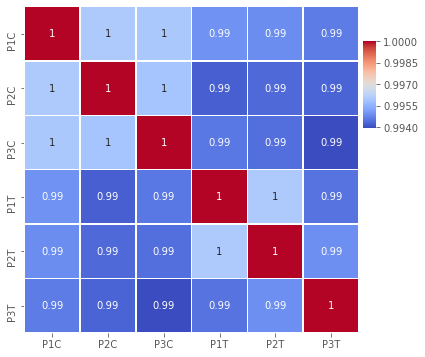
fig,ax=plt.subplots(figsize=(6,6))
cbar_ax = fig.add_axes([.91, .6, .03, .2])
sns.heatmap(np.log2(indata).corr(),
#vmin=-1,
cmap='coolwarm',
annot=True,linewidths=.5,ax=ax, cbar_ax = cbar_ax, cbar=True)
print(ax.get_ylim())
ax.set_ylim(6,0)
plt.savefig('Figures/Figure_2.png')
plt.show()
In [ ]:
QC - MSD¶
In [28]:
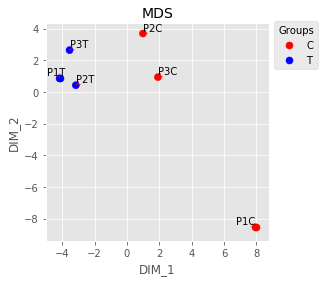
plt.style.use('ggplot')
palette = ['r']*3+['b']*3
fig,ax = plt.subplots(figsize=(4,4), ncols=1, nrows=1)
UT.make_mds(np.log2(indata),palette,ax,color_dictionary={'r':'C','b':'T',
})
plt.savefig('Figures/Figure_3.png')
plt.show()
Compute Length and GC content¶
In [29]:
!mkdir -p InData
In [30]:
!gtf2bed < genomes/tb927_6/tb927_6.gtf > tb927_6.bed
!bedtools nuc -fi genomes/tb927_6/tb927_6.fa -bed tb927_6.bed >InData/GC_content_927.txt
In [31]:
#!bowtie2-build genomes/tb927_5/tb927_5.fa genomes/tb927_5/tb927_5
In [32]:
def get_gene_ids(n):
res = {}
temp = n.split(';')
temp =[n.strip() for n in temp if len(n)>2]
for f in temp:
key = f.split(' ')[0]
value = f.split(' ')[1]
key=key.replace('\"','').replace('\'','').strip()
value=value.replace('\"','').replace('\'','').strip()
res[key]=value
return res['gene_id']
In [33]:
gc_content = pd.read_csv('InData/GC_content_927.txt',sep='\t')
gc_content = gc_content[gc_content['8_usercol']=='transcript']
gc_content['gene_id'] = [get_gene_ids(n) for n in gc_content['10_usercol']]
gc_content = gc_content.drop_duplicates('gene_id')
gc_content.set_index('gene_id',inplace=True)
gc_content=gc_content[['19_seq_len','12_pct_gc']]
gc_content.columns = ['length', 'gccontent']
gc_content.head()
Out[33]:
In [34]:
indata.head()
#indata.astype(int).to_csv('indata.csv',index=True)
Out[34]:
In [35]:
#metadata=pd.DataFrame()
#metadata['samples']=indata.columns
#metadata['treatment']=['C','C','C','D','D','D','DF','DF','DF','F','F','F']
#metadata['batch']=1
#metadata.reset_index().to_csv('metadata.csv')
In [36]:
print(indata.shape)
indata=indata.join(gc_content,how='inner')
gc_content = gc_content[['length', 'gccontent']]
indata.drop(['length', 'gccontent'],axis=1,inplace=True)
In [ ]:
In [ ]:
In [37]:
#indata.loc['mainVSG-427-2']
edgeR to filter low counts¶
In [38]:
%%R -i indata
options(warn=-1)
library("limma")
library("edgeR")
head(indata)
In [40]:
%%R
group <- factor(c(
'C','C','C',
'T','T','T'
))
y <- DGEList(counts=indata,group=group)
keep <- filterByExpr(y)
y <- y[keep,,keep.lib.sizes=FALSE]
counts = y$counts
genes = row.names(y)
In [41]:
%R -o counts,genes
indata = pd.DataFrame(counts,index=genes,columns=indata.columns)
indata.shape
Out[41]:
In [ ]:
In [42]:
indata=indata.join(gc_content,how='inner')
indata.shape
Out[42]:
GC / length content¶
In [43]:
gc_content = indata[['length', 'gccontent']]
indata.drop(['length', 'gccontent'],axis=1,inplace=True)
print(indata.shape,gc_content.shape)
indata.head()
Out[43]:
size factors¶
In [44]:
sizeFactors=indata.sum()
sizeFactors = sizeFactors.values
sizeFactors
Out[44]:
In [45]:
#np.log2(gc_content['length']/1000).plot(kind='hist')
Bias Correction¶
In [47]:
%%R -i gc_content,indata,sizeFactors
library(cqn)
library(scales)
In [48]:
%%R
stopifnot(all(rownames(indata) == rownames(gc_content)))
cqn.subset <- cqn(indata, lengths = gc_content$length,
x = gc_content$gccontent, sizeFactors = sizeFactors,
verbose = TRUE)
In [49]:
#%R cqn.subset
Viz Bias¶
In [50]:
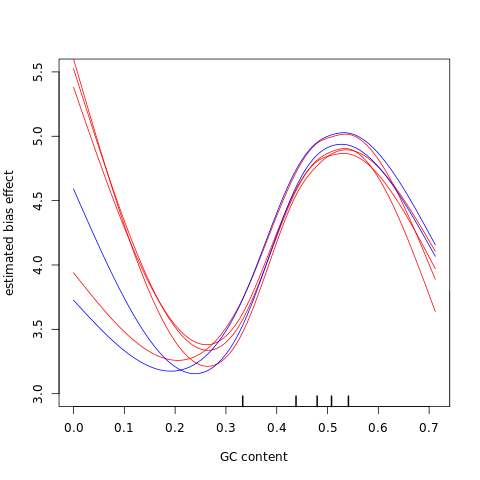
%%R
cqnplot <- function(x, n = 1, col = "grey60", ylab="estimated bias effect",
xlab = "", type = "l", lty = 1, ...) {
if(class(x) != "cqn")
stop("'x' needs to be of class 'cqn'")
if(n == 1) {
func <- x$func1
grid <- x$grid1
knots <- x$knots1
}
if(n == 2) {
if(is.null(x$func2))
stop("argument 'x' does not appear to have two smooth functions (component 'func2' is NULL)")
func <- x$func2
grid <- x$grid2
knots <- x$knots2
}
#par(mar=c(5.1, 4.1, 4.1, 8.1), xpd=TRUE)
matplot(replicate(ncol(func), grid), func, ylab = ylab, xlab = xlab, type = type,
col = col, lty = lty, ...)
legend("bottomleft", legend = colnames(x$counts), inset=c(1,0),
title="Samples", lty = lty, col = col)
rug(knots, lwd = 2)
invisible(x)
}
library(repr)
#options(repr.plot.width = 10, repr.plot.height = 0.75)
# Change plot size to 4 x 3
#options(repr.plot.width=4, repr.plot.height=3)
colors <- c(
'red','red','red','red',
'blue','blue','blue',
'green','green','green',
'yellow','yellow','yellow'
)
lty =c(1,1,1,
1,1,1,
1,1,1,
1,1,1)
#png("Figures/Figure_12.png")
#par(mfrow=c(1,2))
cqnplot(cqn.subset, col=colors,
n = 1, xlab = "GC content", lty = lty,
ylim = c(3, 5.5),
)
#dev.off()
#ggsave('plot.png', width=8.27, height= 11.69) #A4 size in inches
#dev.off()
In [51]:
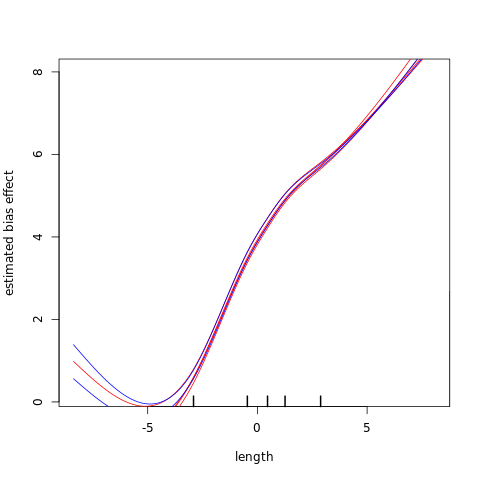
%%R
library(repr)
#options(repr.plot.width = 12, repr.plot.height = 0.75)
# Change plot size to 4 x 3
#options(repr.plot.width=8, repr.plot.height=3)
colors <- c(
'red','red','red','red',
'blue','blue','blue',
'green','green','green',
'yellow','yellow','yellow'
)
lty =c(1,1,1,
1,1,1,
1,1,1,
1,1,1)
#par(mfrow=c(1,2))
#png("Figures/Figure_13.png")
cqnplot(cqn.subset, col=colors,
n = 2, xlab = "length", lty = lty,
ylim = c(0.2,8),
)
#dev.off()
Bias Correction¶
In [52]:
%%R
RPKM.cqn <- cqn.subset$y + cqn.subset$offset
out_table <- RPKM.cqn
head(out_table)
In [53]:
#out_table
In [54]:
%R -o out_table
out_table = pd.DataFrame(out_table,index=indata.index.values,columns=indata.columns)
out_table.head()
Out[54]:
Visualise Normalized Counts¶
In [55]:
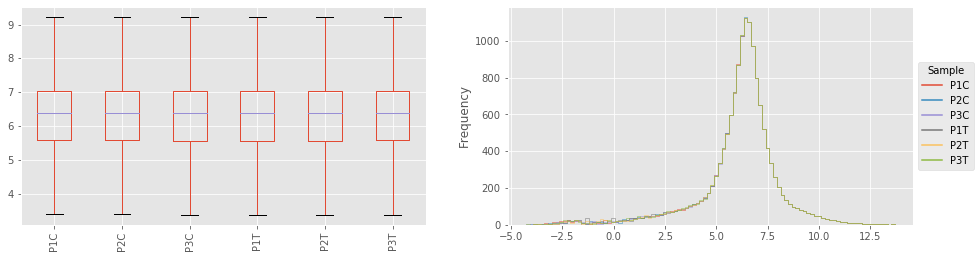
fig,axes=plt.subplots(figsize=(16,4),ncols=2)
ax = axes[0]
out_table.plot(kind='box',ax=ax,rot=90,showfliers=False)
ax = axes[1]
out_table.replace(-np.inf,-1.5).plot(kind='hist',
histtype='step',
bins=100,ax=ax)
UT.hist_legend(ax,'Sample')
#ax.set_xticklabels(out_df.columns, rotation=90, )
plt.show()
print(out_table.shape)
Differential Expression Analysis¶
In [72]:
%%R
library(edgeR)
# Make groups
design_with_all <- model.matrix( ~0+group )
y <- DGEList(counts=indata, group = group)
y <- calcNormFactors(y)
y <- estimateDisp(y, design_with_all)
# Estimate dispersion
# Fit counts to model
fit_all <- glmQLFit( y, design_with_all )
In [73]:
%%R
contrastCT <- glmQLFTest( fit_all, contrast=makeContrasts( groupT-groupC, levels=design_with_all ) )
tableCT <- topTags(contrastCT, n=Inf, sort.by = "none", adjust.method="BH")$table
head(tableCT)
In [110]:
%%R
t=topTags(contrastCT,n=20)
t$table
In [111]:
desc_dict['Tb08.27P2.90']
Out[111]:
In [74]:
%R -o tableCT
def mod_table(intable):
intable['mlog10FDR']=-np.log10(intable['FDR'])
intable['mlog10pvalue']=-np.log10(intable['PValue'])
intable['desc']=[desc_dict.get(n,n) for n in intable.index.values]
return intable
tableCT = mod_table(tableCT)
In [114]:
tableCT.sort_values('logFC').tail(10)
Out[114]:
In [ ]:
In [75]:
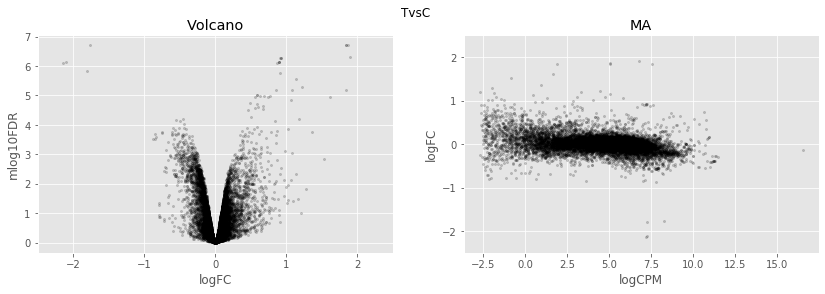
for table,name in zip([tableCT],
['TvsC',]):
fig,axes=plt.subplots(figsize=(14,4), ncols=2, nrows=1)
ax=axes[0]
table.plot(x='logFC',y='mlog10FDR',
kind='scatter',s=5,alpha=0.2,ax=ax,c='black')
ax.set_xlim(-2.5,2.5)
ax.set_title('Volcano')
ax=axes[1]
table.plot(x='logCPM',y='logFC',
kind='scatter',s=5,alpha=0.2,ax=ax,c='black')
ax.set_ylim(-2.5,2.5)
ax.set_title('MA')
plt.suptitle(name)
plt.show()
In [128]:
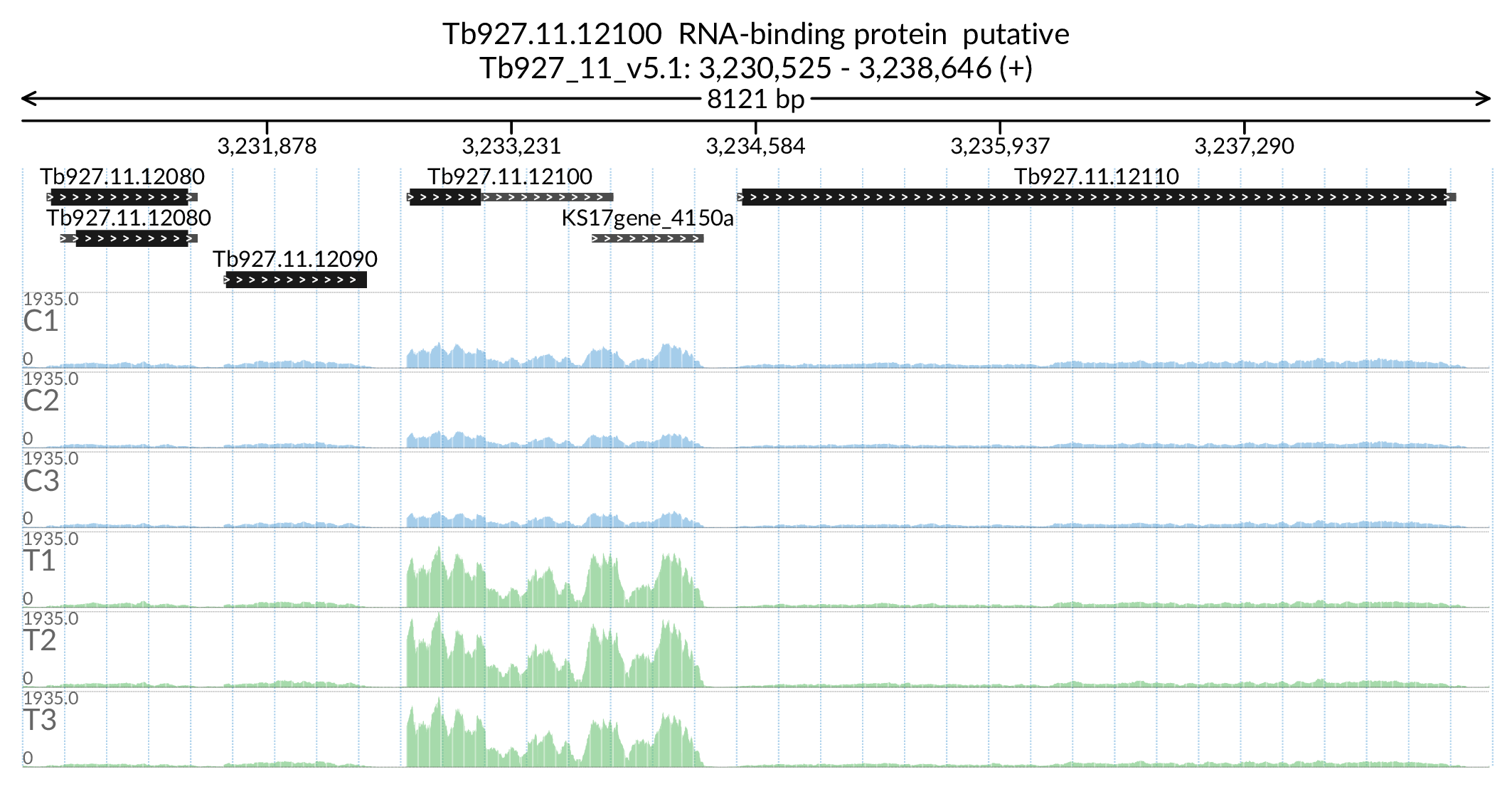
table.loc['Tb927.11.12100']
Out[128]:
In [172]:
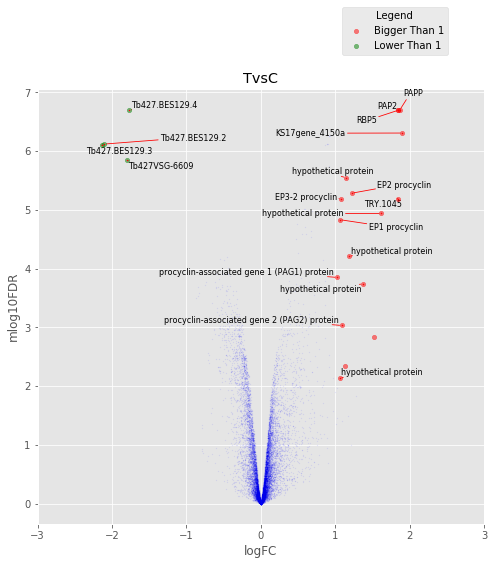
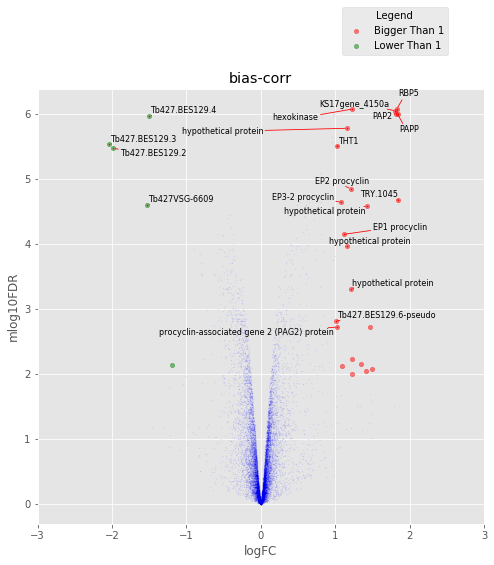
def annotated_volcano(table,name,selection=False):
fig,axes=plt.subplots(figsize=(8,8), ncols=1, nrows=1)
ax=axes
if not selection:
selection = table[((table['logFC']>1)|(table['logFC']<-1))
&(table['mlog10FDR']>2)&(table['logCPM']>0.8)]
else:
selection = table.loc[selection]
annot_index = selection.index.values
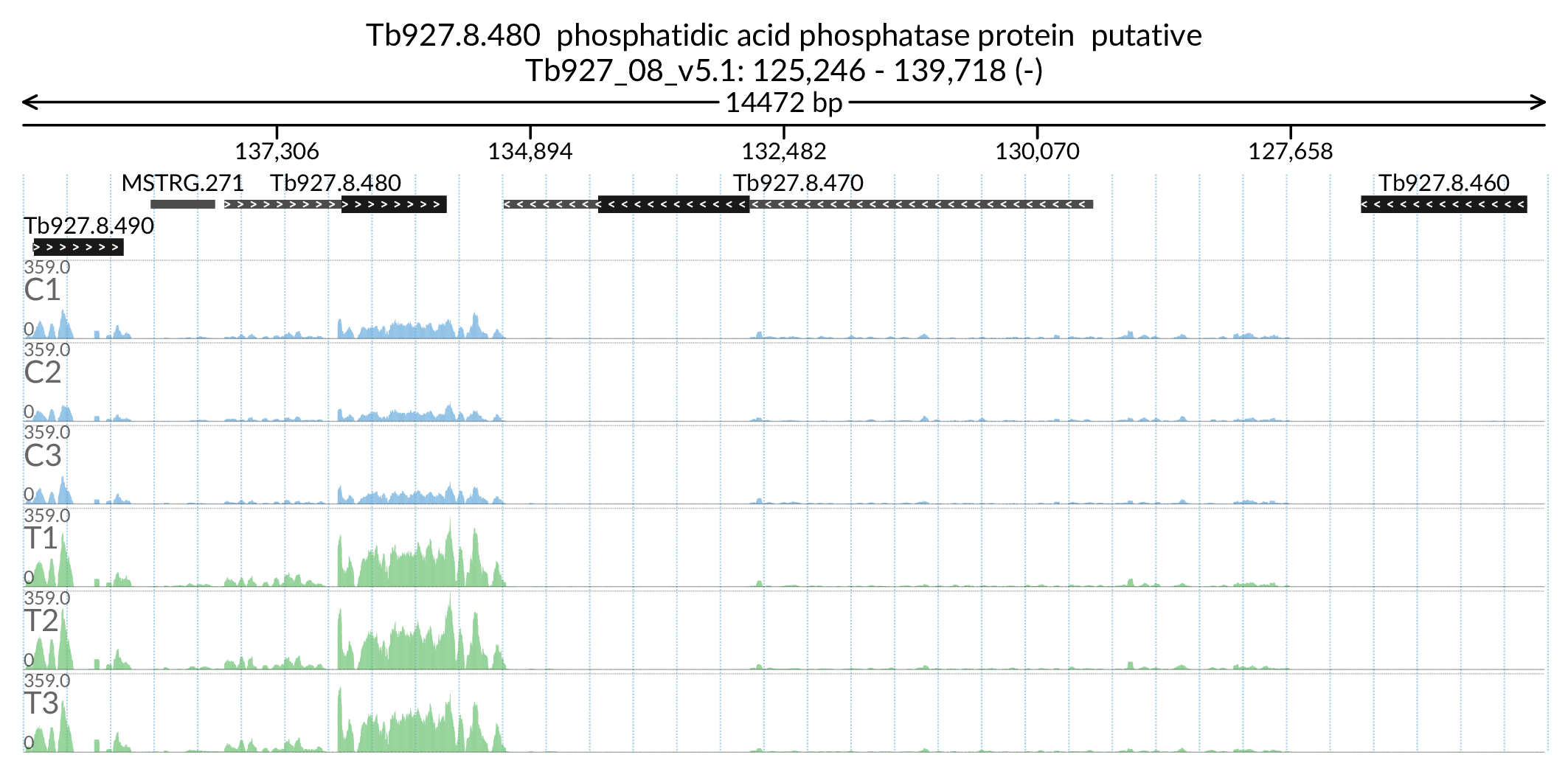
annot_names = selection['desc'].replace({'phosphatidic acid phosphatase protein putative':
'PAPP',
'RNA-binding protein putative':'RBP5',
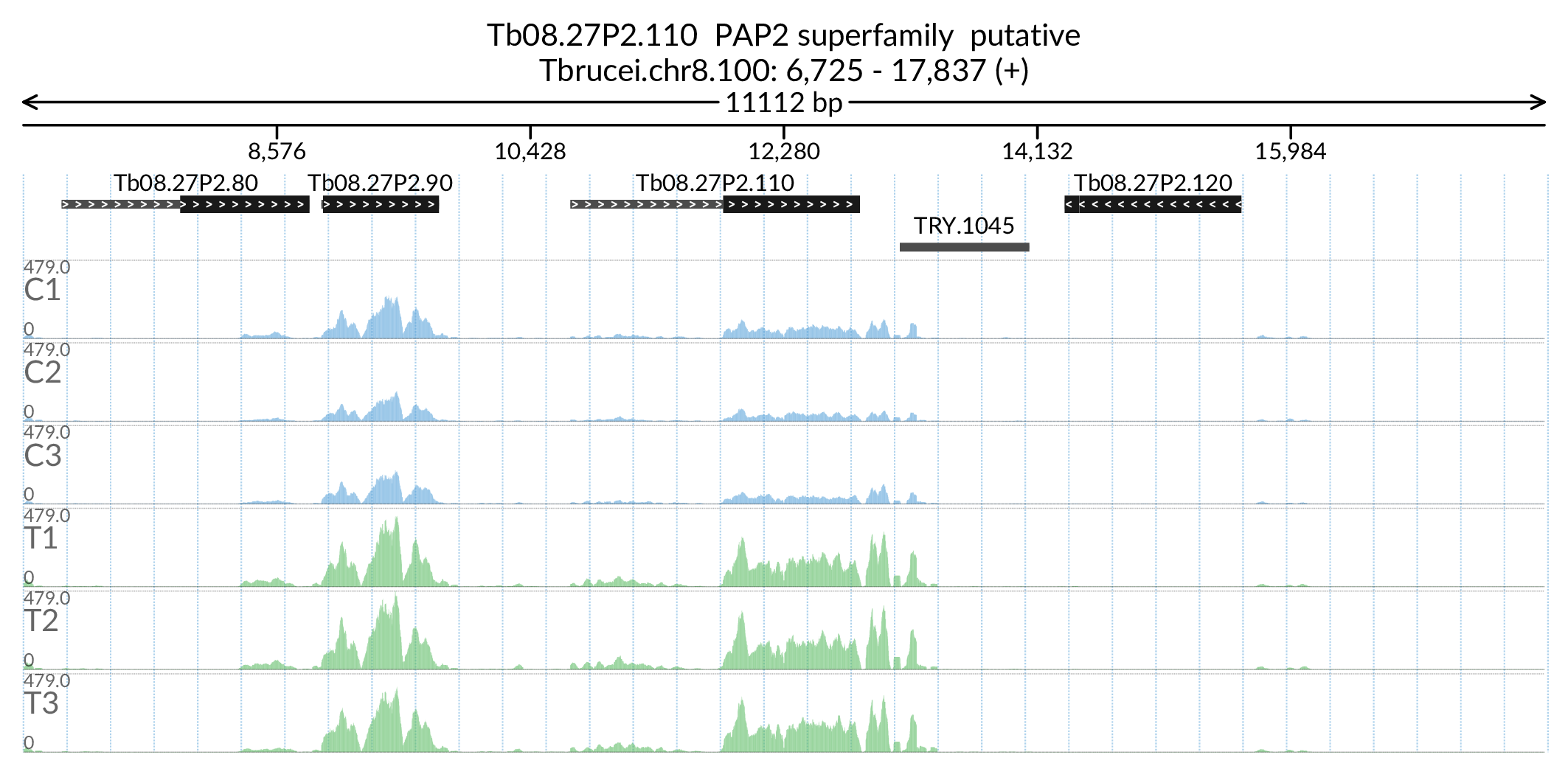
'PAP2 superfamily putative':'PAP2',
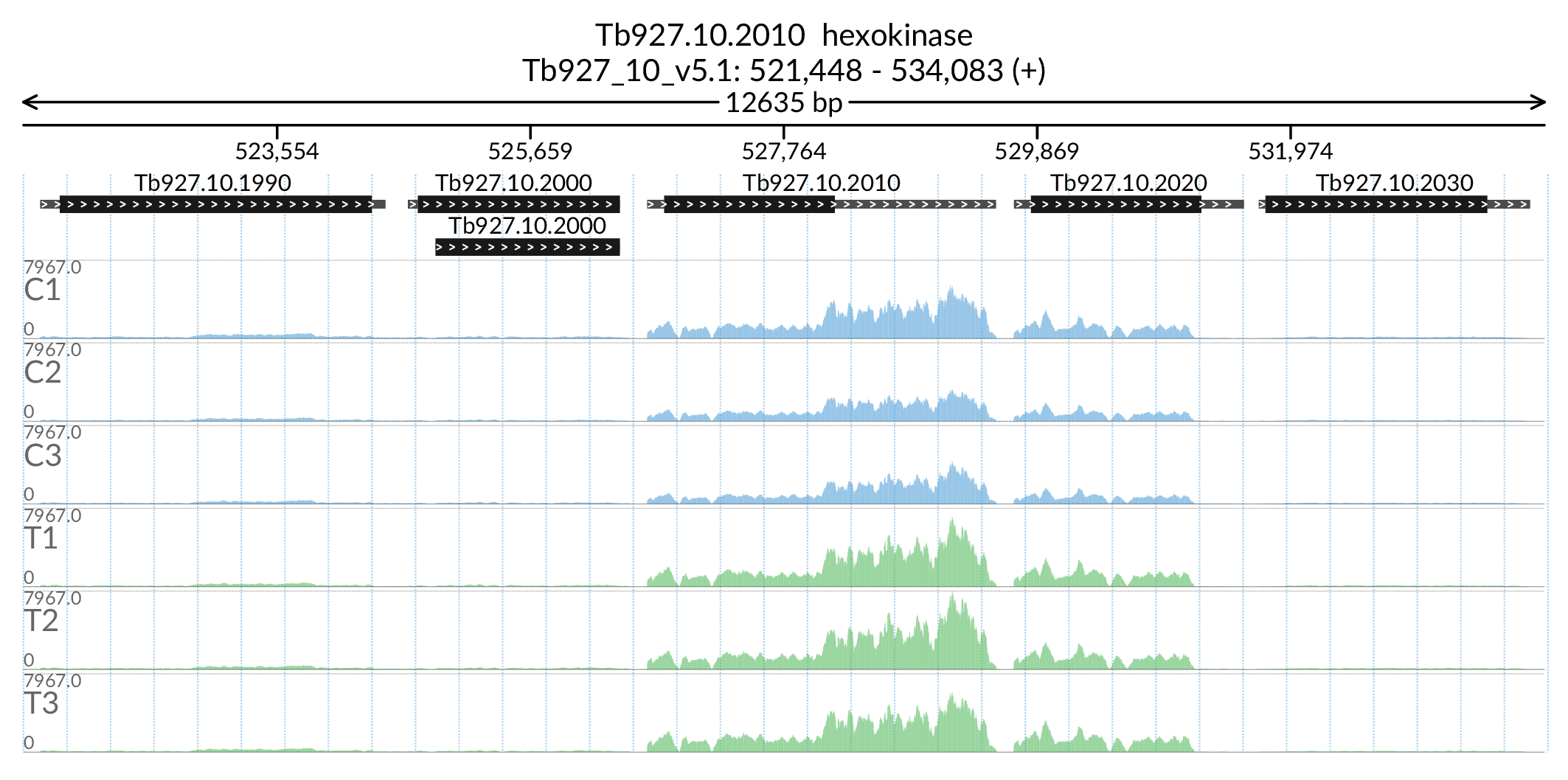
'THT1 - hexose transporter putative':'THT1'})
UT.make_vulcano(
table,
ax,
x='logFC',
y='mlog10FDR',
fc_col = 'logFC',
pval_col = 'FDR',
pval_limit=0.01,
fc_limit=1,
annot_index=annot_index ,
annot_names=annot_names,
expand_text=(1.5, 1.5),
expand_points=(2, 2),)
ax.legend(loc='upper center', bbox_to_anchor=(0.8, 1.2), title='Legend')
ax.set_xlim(-3,3)
plt.title(name)
In [173]:
for table,name in zip([tableCT],
['TvsC',]):
annotated_volcano(table,name)
In [79]:
%%R
#https://rstudio-pubs-static.s3.amazonaws.com/79395_b07ae39ce8124a5c873bd46d6075c137.html
library(edgeR)
# Make groups
design_with_all <- model.matrix( ~0+group )
y <- DGEList(counts=indata,
lib.size = sizeFactors,
group = group,
)
y$offset <- cqn.subset$glm.offset
# Estimate dispersion
y <- estimateGLMCommonDisp( y, design_with_all )
y <- estimateGLMTrendedDisp( y, design_with_all )
y <- estimateGLMTagwiseDisp( y, design_with_all )
# Fit counts to model
fit_all <- glmQLFit( y, design_with_all )
#contrastDC <- glmLRT( fit_all, contrast=makeContrasts( groupD-groupC, levels=design_with_all ) )
#tableDC <- topTags(contrastDC, n=Inf, sort.by = "none", adjust.method="BH")$table
#head(tableDC)
contrastCT2 <- glmQLFTest( fit_all, contrast=makeContrasts( groupT-groupC, levels=design_with_all ) )
tableCT2 <- topTags(contrastCT2, n=Inf, sort.by = "none", adjust.method="BH")$table
head(tableCT2)
In [80]:
%R -o tableCT2
tableCT2 = mod_table(tableCT2)
In [174]:
annotated_volcano(tableCT2,'bias-corr')
In [150]:
for table,name in zip([tableCT2],
['TvsC',]):
fig,axes=plt.subplots(figsize=(14,4), ncols=2, nrows=1)
ax=axes[0]
table.plot(x='logFC',y='mlog10FDR',
kind='scatter',s=5,alpha=0.2,ax=ax,c='black')
ax.set_xlim(-2.5,2.5)
ax.set_title('Volcano')
ax=axes[1]
table.plot(x='logCPM',y='logFC',
kind='scatter',s=5,alpha=0.2,ax=ax,c='black')
ax.set_ylim(-2.5,2.5)
ax.set_title('MA')
plt.suptitle(name)
plt.show()
In [168]:
tableCT2[tableCT2['mlog10FDR']>4].sort_values('logFC').tail(20)
Out[168]:
In [146]:
tableCT2[tableCT2['mlog10FDR']>4].sort_values('logFC').head(5)
Out[146]:
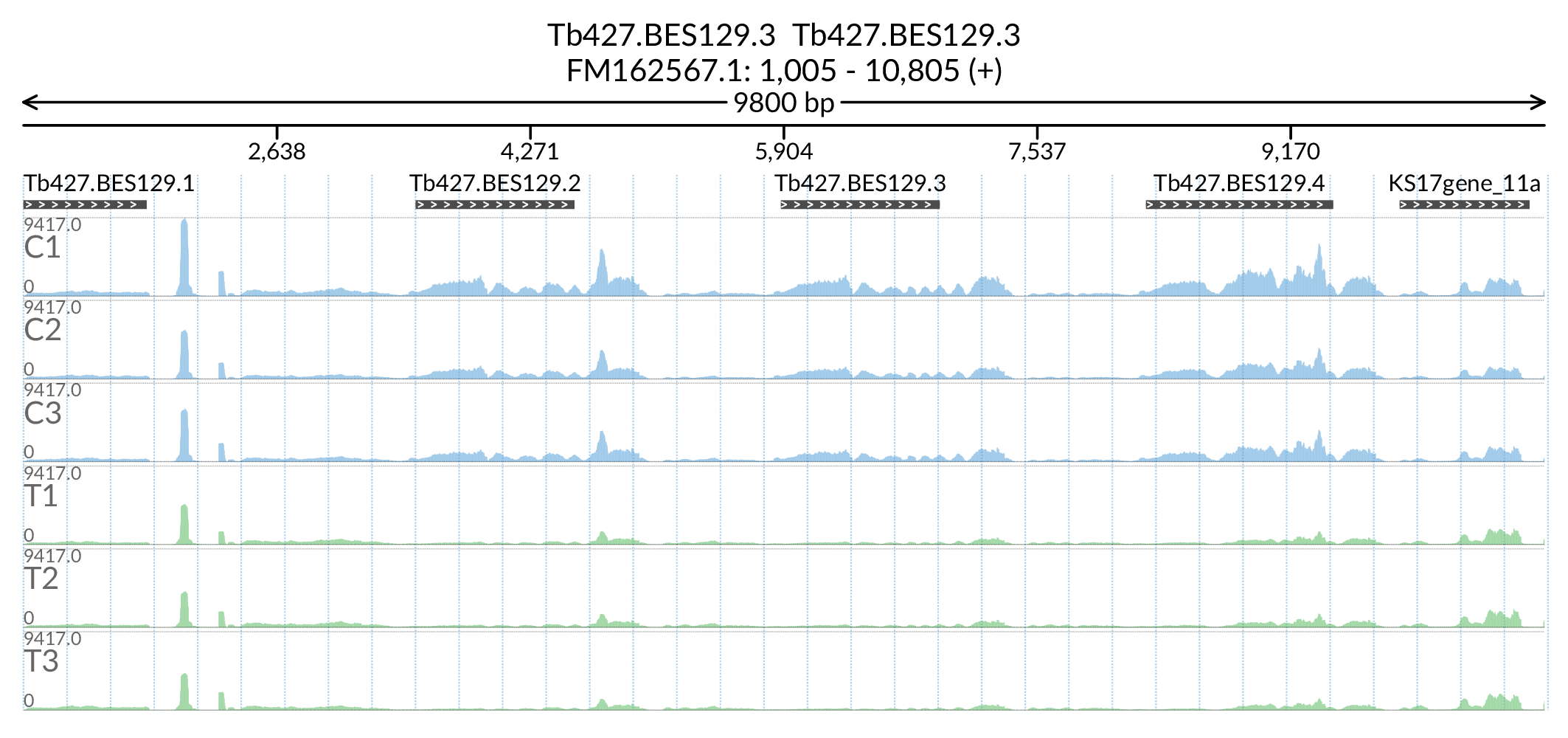
In [177]:
Image('Figures/Tb427.BES129.3_paperFig.png')
Out[177]:
In [178]:
Image('Figures/Tb927.11.12100_paperFig.png')
Out[178]:
In [179]:
Image('Figures/Tb927.8.480_paperFig.png')
Out[179]:
In [180]:
Image('Figures/Tb08.27P2.110_paperFig.png')
Out[180]:
In [181]:
Image('Figures/Tb927.10.2010_paperFig.png')
Out[181]:
In [183]:
!jupyter nbconvert --to html_toc FiguresPaper.ipynb
In [ ]: